
Still-Syndrom
Das Still-Syndrom
Das sowohl bei Kindern als auch bei Erwachsenen auftretende Still-Syndrom ist eine seltene systemische, autoinflammatorische Multiorgan-Krankheit. Es umfasst Systemische juvenile idiopathische Arthritis (SJIA) bei Kindern und Jugendlichen sowie die Erkrankung Adult-Onset Still's Disease (AOSD) bei Erwachsenen.1-4
Informationsmaterial über das Still-Syndrom zum Download finden Sie in unserer Mediathek

Die SJIA ist die seltenste, aber auch schwerste Form der juvenilen idiopathischen Arthritis (JIA) und betrifft nach Experten-Schätzungen ca. 10–20 Prozent der Erkrankungen. Jedes Jahr gibt es 5–20 Neuerkrankungen pro 100.000 Kinder – Jungen und Mädchen erkranken in etwa gleicher Anzahl.1
Die AOSD betrifft Frauen und Männer gleichermaßen und zeigt vorwiegend zwei Erkrankungsgipfel: zwischen dem 15. bis 25. und zwischen dem 36. und 46. Lebensjahr.3-4
AOSD tritt weltweit auf. Es gibt ungefähr 1 Betroffenen pro 100.000 Einwohner, wobei die Dunkelziffer wahrscheinlich relativ hoch ist.2
Typische Symptome des Still-Syndroms sind:
- Wiederkehrende, tägliche Fieberschübe (Remittierendes Fieber),
- Hautausschlag (Exanthem),
- Gelenkentzündung (Arthritis),
- Lymphknotenvergrößerung (Lymphadenopathie),
- Leber- und Milzvergrößerung (Hepatosplenomegalie),
- Entzündung einer serösen Haut, z. B. des Brustfells oder des Herzbeutels (Serositis).
Die Ursachen für das Still-Syndrom sind noch nicht eindeutig wissenschaftlich geklärt
– in zwei Dingen sind sich die Experten jedoch einig:
- Je früher die passende Therapie erfolgt, umso größer ist die Wahrscheinlichkeit der Risikoreduktion für Gelenk-, Organ- und Spätschäden.
- Primäres Therapieziel ist eine schnelle, anhaltende Linderung des akuten systemischen Entzündungsgeschehens.
Daran anschließend gilt es, eine inaktive Erkrankung und eine langfristige Remission zu erreichen sowie Organ- und destruktive Gelenkschäden zu vermeiden.5
Wie bei allen autoinflammatorischen Erkrankungen kommt es auch bei dem Still-Syndrom zu einer Aktivierung des angeborenen Immunsystems. Ausgelöst wird dies höchstwahrscheinlich durch eine Überaktivität von bestimmten Botenstoffen des Immunsystems, den Zytokinen.
Insbesondere bei SJIA spielen die proinflammatorischen Zytokine lnterleukin-1α (IL-1α), lnterleukin-1ß (IL-1ß) und auch Interleukin-6 (IL-6) eine maßgebliche Rolle und „befeuern“ die Aktivität der SJIA. Die Therapien fokussieren sich deshalb auf die Blockade der biologischen Aktivität der Interleukine.6, 7
Verlauf
Die kindliche Form des Still-Syndroms (SJIA) beginnt häufig mit hohem Fieber (über 39 °C) und Hautausschlag (Exanthem). Bei Beginn der Erkrankung ist häufig zunächst keine Gelenkentzündung (Arthritis) erkennbar, doch äußern einige Patienten bereits Gelenkschmerzen. Die Beschwerden können im Verlauf sehr variabel ausgeprägt sein:8, 9, 10
- monozyklisch, ca. 40 % der Patienten,
- polyzyklisch, < 10 %,
- persistierend > 50 % Erkrankung.
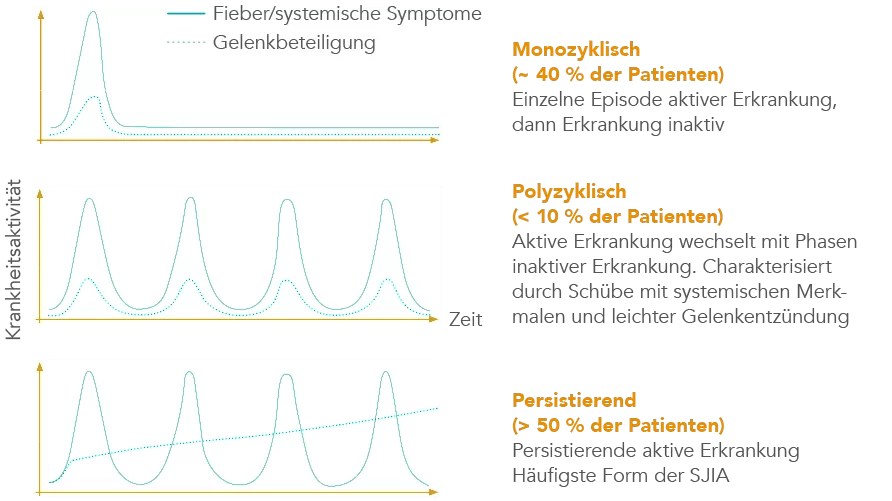
Auch bei Erwachsenen mit dem Still-Syndrom (AOSD) zeigen Patienten unterschiedliche Verläufe. So gibt es Patienten, die nur einen AOSD-Schub haben (20–30 %). Andere Patienten haben immer wieder Schübe (15–45 %) oder eine anhaltende Erkrankung (25–45 %).
Symptome
Die Symptome des Still-Syndroms sind vielseitig, oft wenig spezifisch und zeigen sich je nach Symptom und der systemischen Ausprägung an verschiedensten Stellen des Körpers.
Gemäß der Klassifikation nach ILAR (International League Against Rheumatism) liegt die kindliche Form des Still-Syndroms (SJIA) bei folgenden Kriterien vor:1, 11
- Remittierendes Fieber über mindestens 14 Tage mit 1 bis 2 Fieberspitzen täglich,
- Entzündung in mindestens einem Gelenk, jedoch initial häufig nicht erkennbar (Arthritis),
- Und mindestens eines der folgenden Symptome:
- Flüchtiger Hautausschlag (Exanthem),
- Generalisierte Lymphknotenschwellungen (Lymphadenopathie),
- Leber- und/oder Milzvergrößerung (Hepatomegalie und/oder Splenomegalie),
- Entzündung einer serösen Haut, z. B. des Brustfells oder des Herzbeutels (Serositis).

Auch das Still-Syndrom des Erwachsenen (AOSD) ist eine Ausschlussdiagnose. In der AOSD-Abklärung und Diagnosestellung werden am häufigsten die Klassifikationskriterien von Yamaguchi angewendet. Es müssen bei AOSD fünf Kriterien und davon mindestens zwei Hauptkriterien erfüllt sein:12

Im Rahmen des Still-Syndroms kann es bei allen Patientengruppen zu Komplikationen wie dem Makrophagen-Aktivierungssyndrom (MAS), das etwa bei rund 10 % der betroffenen SJIA-Patienten auftritt, und der Amyloidose kommen.13, 16-18
Bei der Amyloidose bedingt die fortwährende Entzündung im Körper eine ständige Erhöhung der Akute-Phase-Proteine, wie z.B. das Serumamyloid A (SAA). Diese werden in unterschiedlichem Ausmaß als Antwort auf Entzündungsreize gebildet, was zur Schädigung der Nieren führen kann.
Diagnose
Zur Diagnose des Still-Syndroms werden eine umfangreiche Anamnese, Bluttests und Röntgenaufnahmen der betroffenen oder möglicherweise gefährdeten Gelenke vorgenommen.
Liegt ein Still-Syndrom vor, zeigt sich in der Regel folgendes im Labor:1
- BSG stark beschleunigt,
- CRP stark erhöht,
- Erhöhte Anzahl von weißen Blutkörperchen bis 50.000/μl (Leukozytose),
- Erhöhte Anzahl von Blutplättchen (Thrombozytose),
- Anämie (Kinder klagen u. a. über schlechten Allgemeinzustand mit Müdigkeit, Abgeschlagenheit, Appetitlosigkeit),
- Erhöhung von IL-18 und MRP8/14 (S100),
- Eine Überprüfung des Ferritin-Wertes sichert die rechtzeitige Feststellung eines erhöhten MAS-Risikos (> 1.000 ng/ml).
Wichtig zu wissen ist, dass bei dem Still-Syndrom keine antinukleären Antikörper oder Rheumafaktoren nachweisbar sind.1
Informationsmaterial über das Still-Syndrom zum Download finden Sie in unserer Mediathek
Quellen:
- Horneff G. Z Rheumatol. 2010;69(8):719–35 https://www.ncbi.nlm.nih.gov/pubmed/20798949
- Morbus Still (Still-Syndrom des Erwachsenenalters). Deutsche Rheuma-Liga Bundesverband e.V. 3. Auflage 2013. https://www.rheuma-liga.de/fileadmin/user_upload/Dokumente/Mediencenter/Publikationen/Merkblaetter/3.9_Morbus_Still.pdf (zuletzt besucht am 06.12.2023).
- Kedor C et al. Akt Rheumatol. 2017;42:37–45. https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-0042-118879
- Castañeda S et al. Best Pract Res Clin Rheumatol. 2016;30:222–238. https://www.ncbi.nlm.nih.gov/pubmed/27886796
- Professor Dirk Föll, Klinik für Pädiatrische Rheumatologie und Immunologie, Universitäts-klinikum Münster, beim Rheuma-Kongress in Frankfurt / Main https://www.aerztezeitung.de/me-dizin/med_specials/special-knochen-gelenke/article/ 927590/still-syndrom-konsens-protokolle-therapie.html
- Vastert SJ et al. Arthritis Rheumatol. 2014;66(4):1034–43.
- Niehues T. S2-Therapieleitlinie der Juvenilen Idiopatischen Arthritis (2. Auflage, Stand: 10/2011)
- Woo P. Nat Clin Pract Rheumatol. 2006;2(1):28–34 https://www.ncbi.nlm.nih.gov/pubmed/16932649
- Mellins ED et al. Nat Rev Rheumatol. 2011;7(7):416–26 https://www.ncbi.nlm.nih.gov/pubmed/21647204
- Singh-Grewal D et al. Arthritis Rheum. 2006;54:1595–1601 https://onlinelibrary.wiley.com/doi/full/10.1002/art.21774
- Petty RE et al. J Rheumatol. 2004;31(2):390–2 https://www.ncbi.nlm.nih.gov/pubmed/14760812
- Yamaguchi-Klassifikations-kriterien (1992)
- Ravelli A et al.. Lancet 2007; 369: 767–78.
- Frosch M et al. Rheumatology 2008; 47(2):121–5.
- Gurion R et al. Int J Inflam. 2012; 2012: 271569.
- Weiss JE, Ilowite NT: Juvenile idiopathic arthritis. Pediatr Clin North Am 2005; 52(2):413–42, vi.
- Sawhney S, Woo P, Murray K: Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child 2001; 85: 421–6
- Stephan JL, Kone-Paut I, Ga- lambrun C et al.: Reactive Haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology (Oxford) 2001; 40: 1285–92.
- Feist E et al. Nat Rev Rheumatol. 2018,14:603–618.
- Hinze et al. Pediatric Rheumatol Online J.2018;16:7
- Vastert B et al. Pediatric Rheumatol. 2018, 16(Suppl 2):O08