
Familäres Mittelmeerfieber (FMF)
Das Familiäre Mittelmeerfieber (FMF)
Das Familiäre Mittelmeerfieber (FMF) ist eine seltene autoinflammatorische Erkrankung. Es wird in der Regel autosomal-rezessiv vererbt. Die Betroffenen leiden unter immer wiederkehrenden Fieberattacken und damit verbundenen Schmerzen, insbesondere im Brust- und Bauchraum oder in den Gelenken.3 Die Krankheitsschübe setzen meist bereits in der Kindheit ein.1
Informationsmaterial über das FMF zum Download finden Sie in unserer Mediathek
Epidemiologie/Verbreitung
FMF ist eine seltene Erkrankung und kommt gehäuft bei Menschen mit Abstammung aus dem Mittelmeerraum oder aus dem Nahen Osten mit arabischer, armenischer, jüdischer (insbesondere nicht-aschkenasischer), türkischer oder italienischer Herkunft vor. Weltweit sind etwa 150.000 Menschen betroffen.4 Die Zahl der Merkmalsträger, d. h. derjenigen, die den für die Erkrankung verantwortlichen Gendefekt in sich tragen, ohne zu erkranken, dürfte jedoch wesentlich höher sein. Durch die weltweite Migration ist eine steigende Häufigkeit/Prävalenz der Erkrankung in Westeuropa, USA und Australien zu beobachten.
In der Gesamtbevölkerung in Deutschland bei Kindern bis 16 Jahren beträgt die Prävalenz ca. 0,005 %. Um das Jahr 2012 waren insgesamt ca. 4.000 Fälle in Deutschland bekannt.5 Bei der türkischstämmigen Bevölkerung bis 16 Jahre betrug die Prävalenz nach vorsichtigen Schätzungen damals etwa 0,1 %.6 Es sind jedoch keine Gesamtzahlen zu FMF in Deutschland bekannt.
Ursachen
Auslöser für das Familiäre Mittelmeerfieber ist in der Regel eine Mutation im sogenannten MEVF (Mediterranean Fever)-Gen. Es befindet sich auf dem Chromosom 16 und kodiert für das Protein Pyrin.
Man hat ursprünglich angenommen, dass FMF ausschließlich klassisch autosomal-rezessiv vererbt wird. Dies bedeutet, dass der Gendefekt auf beiden Allelen vorhanden sein muss, damit die mit FMF verbundenen unkontrollierten Entzündungen im Körper entstehen.
Heute weiß man jedoch, dass bei bis zu einem Fünftel der Betroffenen nur eine (heterozygote) oder keine Mutation im MEFV-Gen vorliegt, weshalb man davon ausgeht, dass es noch andere genetische Ursachen für die Erkrankung gibt.
Andererseits kommt es auch vor, dass Menschen mit zwei Mutationen im MEFV-Gen nie erkranken oder aber als gefährliche Spätfolge eine Amlyoidose entwickeln. Genaue Zahlen zur Häufigkeit gibt es hierzu jedoch nicht.7
Symptome
Die typischen Symptome11-13 sind (Abb. 1):
- Rezidivierende (wiederkehrende) kurze Fieberepisoden (Dauer: 1-3 Tage).
- Hohes Fieber, oftmals bis 40°C – dies kann in der frühen Kindheit manchmal auch das einzige Symptom sein.
- Schmerzen in Bauch, Brust oder Gelenken, Gelenkentzündungen/-schwellungen (Synovitis, z.B. am Knie, Fuß- oder Handgelenk), verursacht durch Polyserositis (Pleuritis, Peritonitis, selten Perikarditis) und Arthritis.
- Schwere, schmerzhafte Hautrötungen (erysipelartiges Erythem), häufig im Bereich der Füße und/oder der Unterschenkel.
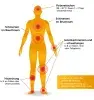
Abb. 1: Typische Symptome des Familiären Mittelmeerfiebers
Verlauf
Die Art, Schwere und Dauer der Symptome und Krankheitsschübe variiert stark von Patient zu Patient, aber auch beim Patienten selbst von Schub zu Schub. Zwischen den einzelnen Fieberattacken sind die Betroffenen in der Regel symptomfrei.
Etwa 90 % der Betroffenen erkranken vor dem 20. Lebensjahr, davon 2/3 bereits vor dem 10. Lebensjahr.6
Bei einem frühen Erkrankungsalter, also zwischen dem 2. und 7. Lebensjahr werden häufig schwere Verläufe beobachtet.1
Es wurden auch Fälle beschrieben, bei denen die Erkrankung erst nach dem 40. (oder sogar nach dem 60.) Lebensjahr auftrat.8
Die Auswirkungen von FMF sind bei Kindern und Erwachsenen ähnlich, allerdings treten einige Symptome wie etwa Arthritis (Gelenkentzündung) oder Myosistis (Entzündung der Muskulatur) häufiger in der Kindheit auf.
Wird die Erkrankung nicht gut kontrolliert, kann sich eine Amlyidose entwickeln. Bei dieser speziellen, durch FMF verursachten Form der Amyloidose lagern sich unlösliche Proteine in den Nieren ab, was auf Dauer zu schweren Nierenschäden bis hin zum Nierenversagen führen kann.6
Es gibt drei Verlaufsformen des FMF:9
- Typ I: Hierbei handelt es sich um die klassische Form mit den oben beschriebenen Symptomen.
- Typ II: Die Amyloidose wird als „Erstmanifestation“ der Erkrankung festgestellt. Die typischen Fieberattacken treten zeitlich nach Auftreten der Amyloidose auf oder die Amyloidose bleibt das einzige Symptom der Erkrankung.
- Typ III: Trotz Vorhandensein von zwei Mutationen im MEVF-Gen treten keine für das FMF typischen Symptome auf.
Diagnose
Die Diagnose des FMF erfolgt klinisch und kann durch eine genetische Untersuchung bestätigt werden.
Hinweise auf ein FMF geben insbesondere das Auftreten kurzanhaltender Fieberattacken, wiederkehrende Schmerzen im Bauchraum, die Abstammung aus der Mittelmeerregion, erhöhte Entzündungsparameter sowie Anzeichen einer Serositis (Peritonitis, Pleuritis, Synovitis).
Um die Diagnose mithilfe verschiedener Testmethoden sicher stellen zu können, sollte der Arzt während eines Fieberschubes aufgesucht werden. Dazu gehören:
- Blutuntersuchung: Bestimmung der Konzentration des C-reaktiven Proteins (CRP) und der Blutsenkungsgeschwindigkeit (BSG) während des Schubes und im Nachgang.
- Urinuntersuchung: Bedeutsam für Amyloid-Diagnostik. Der Urin wird auf Vorliegen einer Proteinurie untersucht.
- Nierenbiopsie: Entnahme von kleinen Gewebeproben aus der Niere zur Labor-Untersuchung für einen Amyloidose-Nachweis.
Spricht der Patient auf Colchicin an, gilt dies als wichtiger Hinweis auf das Vorliegen eines FMF.
Zur Untermauerung der Diagnose kann eine Genanalyse durchgeführt werden. Sie zeigt an, ob der Patient das mutierte MEFV-Gen in sich trägt.
Informationsmaterial über das FMF zum Download finden Sie in unserer Mediathek
Quellen:
- Hitzegrad A, Kallinich T. Akt Rheuma 2017; 42(1): 46-52
- Samuels J, Ozen S. CurrOpinRheumatol 2006; 18: 108–117
- Köhler BM, Lorenz H-M, Blank N. Eur J Rheumatol 2018; 5(4): 230-234
- Wang DQH et al. J Genet Syndr Gene Ther 2014; 5: 5, https://www.longdom.org/open-access/familial-mediterranean-fever-from-pathogenesis-to-treatment-2157-7412-5-248.pdf, aufgerufen am 06.12.2023
- Lainka E et al. Rheumatol 2012; 32: 3253-3260
- Hitzegrad A. Familiäres-Mittelmeerfieber-Patienten mit heterozygotem Genotyp: Eine Patientengruppe mit einem milderen Verlaufsprofil, Dissertation 9/2018
- Kallinich T. arthritis + rheuma 2013; 6: 379-385 https://www.gkjr.de/wp-content/uploads/2019/10/fmf_kallinich_ar_2013-33-6_20535.pdf, aufgerufen am 06.12.2023
- Ricci P et al. Clin Rheumatol 2020; 39 (2): 585–594
- Kallinich T. Natürlicher Krankheitsverlauf und klinische Manifestationen. In: Kallinich T, Wittkowski H, editors. Familiäres Mittelmeerfieber. 1 ed. Bremen: UNI-MED; 2014. p. 51-62
- Livneh A et al. Arthritis Rheum 1997; 40 1879-1885
- Kallinich T et al. Z Rheumatol 2019; 78: 91-101
- Oezen, S, Batu ED, Demir S. Front Immunol 2017; 8: 253
- Fachinformation Kineret® (Anakinra), aktueller Stand